
Auf der Suche nach Rückständen von Pflanzenschutzmitteln in Bienen, Pflanzen und Bienenprodukten
Looking for residues of pesticides in bees, plants and bee products
Journal für Kulturpflanzen, 72 (5). S. 141–153, 2020, ISSN 1867-0911, DOI: 10.5073/JfK.2020.05.04, Verlag Eugen Ulmer KG, Stuttgart
 Dies ist ein Open-Access-Artikel, der unter den Bedingungen der Creative Commons Namensnennung 4.0 International Lizenz (CC BY 4.0) zur Verfügung gestellt wird (https://creativecommons.org/licenses/by/4.0/deed.de).
Dies ist ein Open-Access-Artikel, der unter den Bedingungen der Creative Commons Namensnennung 4.0 International Lizenz (CC BY 4.0) zur Verfügung gestellt wird (https://creativecommons.org/licenses/by/4.0/deed.de).Die chemisch-analytische Bestimmung von Schadstoff-Rückständen verschiedenen Ursprungs ist essentiell für die Expositionsermittlung im Rahmen der Aufklärung von Bienenvergiftungen und von Studien zur Bewertung des Risikos für Bienen. Die etablierte und umfassend validierte Multimethode ist sowohl zur Bestimmung von Rückständen in Bienen- und Pflanzenproben als auch für diverse Bienenprodukte (z.B. Pollen/Bienenbrot, Gelée Royal oder Wachs) sehr gut geeignet. Die Zusatzversuche zur Methodenüberprüfung wurden mit dem jeweils aktuellen Wirkstoffspektrum der Untersuchungsstelle für Bienenvergiftungen und verschiedenen Zusatzkonzentrationen durchgeführt. Die Zusatzkonzentrationen wurden so gewählt, dass möglichst alle Wirkstoffe des Untersuchungsprogramms sowohl mit LC‑MS/MS als auch mit GC‑MS bestimmt werden konnten. Mit den Matrices „Bienen“ und „Raps“ wurden Validierungen mit den Konzentration 1, 10 und 50 μg/kg durchgeführt. Die Quantifizierung wurde mit Matrix-Standards vorgenommen, um die durch Probeninhaltsstoffe hervorgerufenen Matrixeinflüsse zu reduzieren.
Für 240 Wirkstoffe (vor allem Insektizide, Fungizide), die 2011 im Screening-Programm enthalten waren, lagen die Wiederfindungsraten mit Bienenmatrix beim Zusatzniveau von 10 μg/kg überwiegend zwischen 70% und 110%, mit relativen Standardabweichungen unter 15%.
Stichwörter: Rückstandsanalytik, Multimethode, Pestizide, LC‑MS/MS, GC‑MS, Bienen, Pflanzen
The chemical-analytical determination of pollutant residues of various origins is essential for the determination of exposure in the context of the investigation of bee poisoning and of studies to assess the risk to bees. The established and extensively validated multi-method is very well suited for the determination of residues in bee and plant samples as well as for various bee products (e.g. pollen/bee bread, royal jelly or wax). The validation experiments were carried out with the current spectrum of active substances of the investigation center for bee poisoning incidents and various concentration levels. The concentrations were chosen so that as far as possible all active substances of the investigation program could be determined with LC-MS/MS and GC-MS. Validations with the concentrations 1, 10 and 50 μg/kg were carried out with the matrices “bees” and “rape”. The quantification was carried out with matrix standards in order to reduce the matrix influences caused by sample ingredients. For 240 active substances (especially insecticides, fungicides), included in the screening program in 2011, the recovery rates with the bee matrix at the concentration level of 10 μg/kg were mostly between 70% and 110%, with relative standard deviations below 15%.
Key words: Residue analysis, multi-method, pesticides, LC-MS/MS, GC-MS, bees, plants
Bestäuberinsekten sind ein bedeutsamer Bestandteil der globalen Artenvielfalt. Etwa 80 Prozent aller Wild- und Nutzpflanzen in Deutschland sind auf die Bestäubung von Insekten angewiesen. Seit vielen Jahren werden Rückgänge insbesondere bei Wildbienen sowie bei den Pflanzen beobachtet, die auf sie angewiesen sind. Der Verlust an Bestäubern, und damit verbunden der Verlust der Bestäubungsleistung, kann erhebliche negative ökologische und ökonomische Auswirkungen hinsichtlich der Pflanzenproduktion, der Produktqualität, der Erhaltung der Wildpflanzenvielfalt, der Stabilität des Ökosystems und der Ernährungssicherheit haben. Bienen sind verschiedensten Gefährdungen ausgesetzt, die für Schwächung, Verhaltensänderungen, Orientierungs- und Kommunikationsprobleme bis hin zum Tod verantwortlich sein können. Dazu gehören Mangel an ausreichender und vielfältiger Nahrung, Krankheitserreger (Parasiten, Viren), landwirtschaftliche und imkerliche Praktiken, Agrochemikalien, Umwelt- und Lebensraumveränderungen, Klimawandel, gezieltes Vergiften (Frevel) und die mögliche Kombination verschiedener Faktoren (Goulson et al., 2015, Pistorius, 2016, Potts et al., 2010). In neuerer Zeit wurde auch der Einfluss künstlichen Lichts in der Nacht und anthropogener hochfrequenter elektromagnetischer Strahlung auf Bestäuber untersucht (Vanbergen et al., 2019).
Die chemisch-analytische Bestimmung von Schadstoff-Rückständen verschiedenen Ursprungs bei der Aufklärung von Bienenschäden oder als ein Faktor bei der Bewertung des Risikos in verschiedenen Versuchsszenarien ist ein essentieller Bestandteil vieler Untersuchungen zum Bienenschutz (Wisk, 2014, Wernecke et al., 2019).
Das Julius Kühn-Institut (JKI) hat laut Pflanzenschutzgesetz (§ 57 Abs. 2 Nr. 11) die Aufgabe, Bienen auf Schäden durch Pflanzenschutzmittel zu untersuchen. Diese Aufgabe wird von der Untersuchungsstelle für Bienenvergiftungen (UBieV) wahrgenommen, die im Institut für Bienenschutz angesiedelt ist.
Damit ein Schadensverursacher ermittelt werden kann, sollten nicht nur die toten Bienen sondern auch Pflanzen eingesandt werden, an denen sich die Bienen bei der Nahrungssuche vergiftet haben könnten. Eine chemische Untersuchung der eingesandten Proben erfolgt bei geeignetem Probenmaterial in der Regel dann, wenn anhand der vorliegenden Informationen zum Schadensfall und des eingesandten Probenmaterials eine mögliche Vergiftung nicht mit Sicherheit ausgeschlossen werden kann oder zuvor im Biotest (Aedes-Test, Indikator: Larven der Gelbfieber-Mücke Aedes aegypti L.) eine Kontaktgiftwirkung nachgewiesen wurde und somit zu klären ist, ob eine Vergiftung durch Pflanzenschutzmittel vorliegt.
Für die Rückstandsanalytik werden Methoden benötigt, mit denen möglichst viele der hinsichtlich Wasserlöslichkeit, Flüchtigkeit, Molekülgröße und Stabilität sehr unterschiedlichen Substanzen möglichst schnell und einfach in einem Analysengang bestimmt werden können.
Im Jahr 2003 veröffentlichten Klein und Alder eine Multimethode zur Bestimmung von Pflanzenschutzmittelrückständen in Lebensmitteln mit LC‑MS/MS nach Methanolextraktion und Aufreinigung an Diatomeenerde. Es handelt sich dabei um eine schnelle, einfache und effiziente Methode, die in die Amtliche Sammlung von Untersuchungsverfahren nach §§ 64 LFGB (Lebensmittel- und Futtermittelgesetzbuch) aufgenommen wurde (Methode L 00.00–113). Diatomeenerde oder Kieselgur ist für die Rückstandsanalyse in sofort einsetzbaren Kartuschen (z.B. ChemElutTM) erhältlich.
Ebenfalls im Jahr 2003 wurde von Anastassiades et al. eine Extraktionsmethode zur Analyse einer Vielzahl von Pestiziden, die sog. „QuEChERS-Methode“ publiziert. Diese Methode erhielt ihren Namen aufgrund ihrer positiven Eigenschaften im Vergleich mit bis dato eingesetzten Methoden: Quick, Easy, Cheap, Efficient, Rugged, Safe (schnell, einfach, günstig, effizient, robust, sicher) und besteht aus zwei Schritten, Flüssig-Flüssig-Extraktion und Reinigung durch dispersive Festphasenextraktion (dSPE). Die ursprünglich vor allem für fettarme Obst- und Gemüseproben konzipierte Methode wurde in den folgenden Jahren weiterentwickelt, um sowohl zusätzliche Wirkstoffe als auch weitere Probentypen mit sehr verschiedenen Begleitstoffanteilen (die sog. „Matrix“) untersuchen zu können. Die QuEChERS-Methode wurde ebenfalls in die Amtliche Sammlung von Untersuchungsverfahren nach §§ 64 LFGB aufgenommen (Methode L 00.00–115) und ist heute die am meisten verbreitete Methode in der Rückstandsanalytik von Lebensmitteln.
In den folgenden Jahren wurden zunehmend Methoden entwickelt und veröffentlicht, die sich mit der Extraktion organischer Schadstoffrückstände (Pestizide, Tierarzneimittel, Polycyclische Aromatische Kohlenwasserstoffe (PAK), Polychlorierte Biphenyle (PCB), u.a.) aus Honigbienen, Pollen, Wachs und Honig befassen (Przybylski und Segard, 2009, Walorczyk und Gnuskowski, 2009, Wiest et al., 2011, Kasiotis et al., 2014, Al Naggar et al., 2015, Calatayud-Vernich et al., 2016, Kiljanek et al., 2016, Roszko et al., 2016, Tette et al., 2016, Al-Alam et al., 2017, Gil García et al., 2017). Die meisten Methoden basieren auf verschiedenen Modifikationen der QuEChERS-Methode. Diese waren insbesondere für die Analyse fetthaltiger Proben nötig, um den sogenannten „Matrixeffekt“ herabzusetzen. In der analytischen Chemie bezeichnet der Matrixeffekt die Kombination aller Einflüsse auf die Messung und Mengenbestimmung der Zielsubstanz, die durch mitextrahierte, störende Probeninhaltsstoffe hervorgerufen wird (IUPAC).
Für alle Multimethoden gilt, dass die gewonnenen Extrakte, abhängig von den zu bestimmenden Analyten mittels gas- (GC) oder flüssigchromatographischer (LC) Methoden analysiert werden. Als Detektor wird in der Regel ein Massenspektrometer (MS) verwendet, um die gesuchten Substanzen sicher identifizieren zu können.
In der Untersuchungsstelle für Bienenvergiftungen werden die Proben seit Anfang der 1980er Jahre mit rückstandsanalytischen Multimethoden untersucht. Von 1982 bis 2005 wurde für die Extraktion der Bienen- und Pflanzenproben das Lösungsmittelgemisch Hexan/Aceton (3:1, v:v) eingesetzt und die Identifizierung erfolgte mittels GC‑MS-Verfahren. Ab 2006 wurde die von Klein und Alder (2003) publizierte Methode erprobt und nach notwendigen Modifikationen für die Rückstandsanalysen eingesetzt. Seefeld fasste die chemischen Untersuchungen zur Aufklärung von Schadensfällen an Honigbienen durch Pflanzenschutzmittel im Zeitraum 1985 bis 2006 zusammen. Die Publikation erschien im Jahr 2008.
Seit 2007 stehen der UBieV für die Identifizierung und Quantifizierung der Zielsubstanzen in den Probenextrakten neben der GC‑MS- auch LC‑MS/MS-Messsysteme zur Verfügung. Damit wurde der Tatsache Rechnung getragen, dass viele der Pflanzenschutzmittelwirkstoffe vorzugsweise mit der LC‑MS/MS-Technik nachweisbar sind. Daneben gibt es nach wie vor eine Reihe von Wirkstoffen (z.B. chlororganische Insektizide, Pyrethroide), die nur oder besser mit GC‑MS bestimmt werden können. Hinsichtlich der Vielzahl an Substanzen, die mit beiden Messsystemen ermittelt werden können, bietet das Untersuchungsverfahren zusätzliche Sicherheit bei der Klärung von Schadensfällen.
Das zur Schadensfallklärung verwendete Screening-Programm beinhaltet sowohl in Deutschland und anderen europäischen Ländern zugelassene als auch nicht mehr zugelassene Pflanzenschutzmittel-Wirkstoffe, bienentoxische Substanzen aus anderen Anwendungsgebieten (Biozide, z.B. Insektensprays) und solche, die in der Imkerei zum Einsatz kommen. Die dem Screening-Programm zugrundeliegende Multimethode wird kontinuierlich an aktuelle Erfordernisse und neue Wirkstoffe angepasst und umfasst zurzeit ca. 290 Wirkstoffe, etwa die Hälfte sind Insektizide, Akarizide und Varroazide sowie ausgewählte Metaboliten dieser Substanzgruppen. In diesem Artikel wird die Methode im Detail vorgestellt.
Der Ablauf der Rückstandsanalysen ist in Abb. 1 am Beispiel der Schadensfallbearbeitung zusammengefasst. Unterschiede ergeben sich aus der Fragestellung, Art und Menge des für die Rückstandsanalysen zur Verfügung stehenden Probenmaterials. Die Methode wird im Folgenden am Beispiel der Analysen im Schadensfall ausführlich beschrieben. Für die übrigen Probenmaterialien werden nur die wesentlichen Abweichungen vom Grundschema genannt.

Abb. 1. Ablaufschema der Multimethode für die Bestimmung von Rückständen in Bienen- und Pflanzenproben
Die Analysenproben für die Rückstandsanalytik bestehen aus 100 Bienen (ca. 5–10 g) bzw. 5 g Pflanzenmaterial, das vor der Entnahme mit einer Schere vorzerkleinert wird. Für die Bienenprobe werden 10 Bienen gewogen und das Gewicht von 100 Bienen errechnet. Die Analysenprobe wird in ein Zentrifugenglas mit einem Fassungsvermögen von 80 ml eingewogen und nach Zugabe von 50 μl einer Surrogat-Standard-Lösung (Erläuterung unter Punkt „Interne Standards“) mit 20 ml Aceton und einer definierten Menge Reinstwasser (0,055 μS/cm) versetzt. Die Gesamtwassermenge (Restfeuchte in der Probe plus Zugabe) soll bei den Bienenproben 10 ml betragen. Da aufgrund der eingesandten Gesamtprobenmenge in der Regel eine individuelle Wassergehaltsbestimmung nicht möglich ist, erfolgt die Wasserzugabe aufgrund empirisch ermittelter Werte in Abhängigkeit des Gewichtes von 10 Bienen. Bei den Pflanzenproben soll die Gesamtwassermenge 10 ml betragen. Es wird pauschal von einer Restfeuchte von 80% ausgegangen und jeder Analysenprobe werden 6 ml Wasser zugesetzt. Nach einer Einwirkzeit von 30 min werden die Proben 3 min mit einem Dispergiergerät homogenisiert und anschließend 10 min bei 1690 g zentrifugiert. Nach dem Zentrifugieren werden 15 ml des überstehenden Probenextrakts entnommen, mit 5 ml einer 20%igen Natriumchloridlösung versetzt und auf eine ChemElutTM-Kartusche (20 ml, unbuffered, Agilent) gegeben. Nach einer Einwirkzeit von 15 min wird zweimal mit je 50 ml Dichlormethan in einen 250 ml Rundkolben eluiert. Die Eluate werden am Rotationsverdampfer bei max. 35°C Wasserbadtemperatur bis zur Trockne konzentriert. Der Rückstand wird mit 2,5 ml einer Lösung in Acetonitril versetzt, welche die internen, überwiegend isotopenmarkierten Standards (aktuell 15) für die Quantifizierung enthält, und für 30 Sekunden im Ultraschallbad gelöst. Der verschlossene Rundkolben wird über Nacht in einen Tiefkühlschrank (–18°C) gestellt, wodurch störende Begleitstoffe zum Teil ausgefroren werden. Der Probenextrakt wird über einen Spritzenfilter (PTFE 13 mm, 0,2 μm) in ein Vial (Probenfläschchen) filtriert.
Für die Analyse werden 5 g Probe eingewogen und nach Zugabe von 50 μl der Surrogat-Standard-Lösung und 30 ml einer Aceton-Wasser-Mischung (3:1, v:v) ist der weitere Ablauf identisch mit der oben beschriebenen Vorgehensweise für Bienen.
Für die Analyse wird in der Regel 1 g Probe eingewogen. Nach Zugabe einer definierten Menge einer zur Zielstellung passenden Surrogat-Standard-Lösung und 20 ml einer Aceton-Wasser-Mischung (3:1, v:v) ist der weitere Ablauf der Analyse bis zur Konzentrierung des Extrakts identisch mit der oben beschriebenen Vorgehensweise. Die Eluate werden am Rotationsverdampfer bis auf ca. 2 ml konzentriert. Der verbleibende Extrakt wird in ein Reagenzglas mit Normschliff überführt und anschließend mit Stickstoff bei Raumtemperatur zur Trockne eingedampft. Der Rückstand wird mit 1 ml einer Lösung in Acetonitril versetzt, die ausgewählte interne Standards für die Quantifizierung enthält, und für 10 Sekunden im Ultraschallbad gelöst. Nach Lagerung über Nacht im Tiefkühlschrank (–18°C) wird der Probenextrakt über einen Spritzenfilter (PTFE 13 mm, 0,2 μm) in ein Vial filtriert.
Für die Analyse wird 1 g Probe eingewogen. Nach Zugabe einer ausgewählten Surrogat-Standard-Lösung und 20 ml einer Aceton-Wasser-Mischung (2:1, v:v) ist der weitere Ablauf der Analyse bis zur Konzentrierung des Extrakts identisch mit dem oben beschriebenen Prozedere für Bienen und anschließend mit dem für Gelée Royal.
Ein interner Standard (IS) ist eine chemische Verbindung, die der Probe oder dem Probenextrakt in einer bekannten Menge in einem bestimmten Stadium der Analyse zugesetzt wird, um die korrekte Ausführung (eines Teils) der Analysemethode zu überprüfen. Der interne Standard sollte chemisch stabil sein und/oder typischerweise das gleiche Verhalten wie der Zielanalyt zeigen (SANTE, 2019). Der sogenannte Surrogat-Standard (SANTE: prozeduraler interner Standard (P-IS)) ist ein interner Standard, der den Proben zu Beginn der Analyse hinzugefügt wird, um verschiedene Fehlerquellen in allen Phasen der Methode von der Aufarbeitung bis zur Quantifizierung zu erkennen. Die Surrogat-Standard-Lösung enthält drei isotopenmarkierte, in diesen Fällen deuterierte Referenzsubstanzen (Acetamiprid D3, Pirimicarb D6, Chlorpyrifos D10). „Deuteriert“ bedeutet, dass in den betreffenden Ausgangsmolekülen in der Regel mehrere Wasserstoffatome durch Deuterium ersetzt wurden, was mit der Zahl hinter dem „D“ ausgedrückt wird. Diese Substanzen kommen natürlicherweise nicht vor und damit wird sichergestellt, dass die Kontroll-Standards nicht bereits in der zu analysierenden Probe enthalten sind. Sie sind damit Stellvertreter oder Ersatz für die Zielsubstanzen der Analyse. Voraussetzung für die Verwendung von isotopenmarkierten internen Standards ist die massenspektrometrische Messung, die den gleichzeitigen Nachweis der co‑eluierenden, nicht markierten Analyten und der entsprechenden isotopenmarkierten internen Standards ermöglicht. Die im Zuge der Gesamtanalyse für die Surrogat-Standards erzielten Ergebnisse (Wiederfindung) dienen der Kontrolle und werden nicht in die Ergebnisberechnung einbezogen.
Die für die Quantifizierung verwendeten internen Standards werden auch Injektionsstandard (I-IS) oder instrumenteninterner Standard genannt. Diese werden unmittelbar vor dem Bestimmungsschritt (d.h. vor der Injektion) den endgültigen Extrakten hinzugefügt. Dies ermöglicht eine Überprüfung und mögliche Korrektur von Schwankungen des Einspritzvolumens (SANTE, 2019) und der Messempfindlichkeit. Letzteres gilt vor allem für die GC‑MS.
Für die Identifizierung und Quantifizierung der Zielsubstanzen in den Probenextrakten stehen ein GC‑MS- (DSQTM II, Thermo Electron Corporation), ein GC‑MS/MS- (TSQTM 8000 Evo, Thermo Scientific) und zwei LC‑MS/MS-Messgeräte (4000 QTRAP®, AB/MDS SCIEX und QTRAP® 6500+, SCIEX) zur Verfügung.
Im Folgenden werden die Mess- und Auswerteparameter für die Gerätesysteme (DSQTM II, 4000 QTRAP®) detailliert beschrieben, die in der Regel im Rahmen von Analysen eingesetzt werden, bei denen das gesamte Untersuchungsspektrum (z.B. Schadensfallklärung) gefragt ist. Die beiden anderen massenspektrometrischen Gerätesysteme werden in der Regel für Analysen im Rahmen von Projekten eingesetzt. In diesen Fällen sind die zu analysierenden Wirkstoffe bekannt und die Messbedingungen werden für die jeweilige Fragestellung optimiert.
Für die GC‑MS-Messungen wird ein DSQTM II‑Gerät (Single Stage Quadrupol) gekoppelt an einen Trace GC UltraTM mit split/splitless-Injektor (210°C, Splitless: 0,0 bis 1,5 min) und CTC CombiPAL Autosampler (CTC Analytics) (10°C) in den Messmodi EI und NICI verwendet. Die Trennung erfolgt an einer ZebronTM ZB-MultiResidueTM-1 Kapillare (30 m, 0,25 mm i.D., 0,25 μm Filmdicke, Phenomenex) mit Helium 5.0 als Trägergas, einem Fluss von 1,2 ml/min (constant flow) und folgendem Temperaturprogramm: 70°C, 2 min halten, mit 5°C/min auf 320°C heizen, 10 min halten.
Die massenspektrometrischen Messbedingungen sind im EI‑Modus: –70 eV, Full scan 50–650 u, Ionenquelle 225°C, Transferline 275°C und im NICI‑Modus: –70 eV, Full scan 20–650 u, CI Gas Methan 1,7 ml/min, Ionenquelle 180°C, Transferline 275°C.
Das Injektionsvolumen ist 1 μl und die Messzeit beträgt 62 min. Zwischen den Proben wird Acetonitril injiziert und mit einem kurzen Temperaturprogramm gemessen.
Als Steuerungs- und Auswerte-Software wird XcaliburTM 1.4 SR 1 genutzt.
Die Zielsubstanzen werden nach der chromatographischen Trennung und massenspektrometrischer Detektion in zwei Schritten anhand ihrer Retentionszeit und der Full Scan‑Spektren identifiziert. Das Full Scan-Spektrum zeigt die Gesamtheit aller zum Messzeitpunkt gebildeten und detektierten Ionen des definierten Massenbereichs (50–650 u bzw. 20–650 u)). Im halbautomatischen Grob‑Screening wird anhand von Layouts (Abb. 2) eine Liste der potentiell in der Probe enthaltenen Wirkstoffe erstellt, deren Identität im Fein‑Screening durch Spektrenvergleich abgesichert wird. Für die Identifizierung werden vorrangig die in eigenen EI- und NICI‑Bibliotheken niedergelegten Substanzspektren herangezogen.
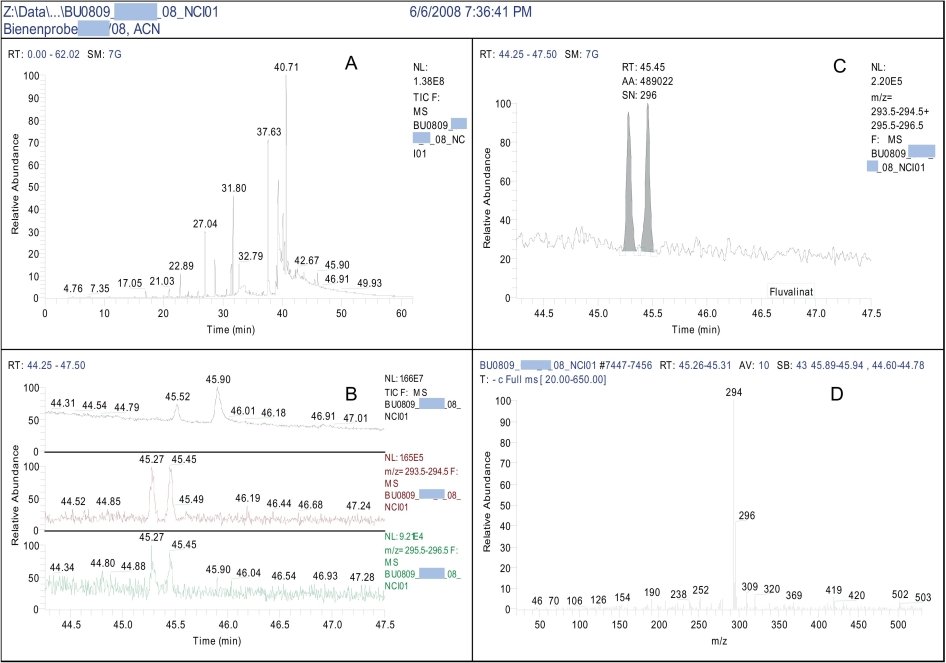
Abb. 2. Layouts zur Auswertung der GC-MS-NICI-Messergebnisse am Beispiel von tau-Fluvalinat in einer Bienenprobe. Chromatogramm (A), Fragment-Ionen für Grob-Screening (B) und Quantifizierung (C), Spektrum für Bibliotheksvergleich (D)
Für die LC‑MS/MS-Messungen kommt ein Triple Stage Quadrupole Massenspektrometer 4000 QTRAP® mit ESI-Quelle (Elektro-Spray-Ionisation) zum Einsatz, das an eine Prominence UFLC XR HPLC-Anlage (Shimadzu) gekoppelt ist. Es werden Messungen mit positiver (Positiv-Modus) und negativer Ionisation (Negativ-Modus) durchgeführt. Die Säulenofentemperatur ist auf 40°C und die des Autosamplers auf 15°C gesetzt.
Im Positiv‑Modus 1 werden die Messungen an einer SynergiTM Hydro Trennsäule (150 × 3 mm; 4 μm; 80 Å) mit einer SecurityGuard cartridge AQ C18 als Vorsäule (4 × 2,0 mm) (beide von Phenomenex) durchgeführt. Es werden 10 μl Probenextrakt injiziert, und die Messzeit umfasst 22 Minuten.
Im Negativ‑Modus wird eine Kinetex® C18-Trennsäule (50 × 3 mm; 2,6 μm; 100 Å) mit einer SecurityGuard Cartridge Ultra als Vorsäule verwendet (beide von Phenomenex). Es werden 5 μl Probenextrakt injiziert und die Messzeit umfasst 6 Minuten.
Diese Trennsäule wird auch für die Messungen im Positiv-Modus 2 verwendet, um große Moleküle wie Azadirachtin und Abamectin bestimmen zu können. Es werden ebenfalls 5 μl Extrakt injiziert und die Messung dauert 8 min.
In allen Mess-Modi wird zwischen den Proben Acetonitril injiziert.
Für die Gradiententrennung werden in allen Fällen ein Methanol/Wasser-Gemisch (90:10, v:v, Solvent A) und Wasser (Solvent B) verwendet, die jeweils 0,1% Essigsäure und 5 mM Ammoniumformiat enthalten. Die jeweiligen Flussraten und Gradienten sind in der folgenden Tab. 1 zusammengefasst.
Tab. 1. Für die Trennung verwendete Gradienten in der LC-MS/MS
Gradient im: | Zeit | Fluss | Solvent A | Solvent B |
Positiv-Modus 1 | 0,0 | 0,6 | 10 | 90 |
| 12,0 | 0,6 | 100 | 0 |
| 18,0 | 0,6 | 100 | 0 |
| 18,1 | 0,6 | 10 | 90 |
| 22,0 | 0,6 | 10 | 90 |
Negativ-Modus | 0,0 | 0,7 | 10 | 90 |
| 0,5 | 0,7 | 100 | 0 |
| 3,8 | 0,7 | 100 | 0 |
| 4,8 | 0,7 | 10 | 90 |
| 6,0 | 0,7 | 10 | 90 |
Positiv-Modus 2 | 0,0 | 0,9 | 10 | 90 |
| 3,0 | 0,9 | 100 | 0 |
| 6,0 | 0,9 | 100 | 0 |
| 6,1 | 0,9 | 10 | 90 |
| 8,0 | 0,9 | 10 | 90 |
Die massenspektrometrischen Parameter sind wie folgt gewählt: Scan type: sMRM, Quellentemperatur: 500°C, Ion spray voltage: +5500 V im Positiv‑Modus 1 und 2 sowie –4500 V im Negativ‑Modus, Curtain Gas (N2): 35 psi, Ion Source Gas 1 (Nebulizer Gas, N2): 70 psi, Ion Source Gas 2 (Turbo Gas): 50 psi, Collision gas (N2): high. Als Steuerungssoftware wird Analyst® in der Version 1.5.2 und für die Identifizierung und Quantifizierung die Software MultiQuantTM in der Version 2.1.1 verwendet.
Die Zielsubstanzen werden anhand ihrer Retentionszeit und dreier charakteristischer MRM-Übergänge identifiziert. Abb. 3 zeigt dies am Beispiel einer Bienenprobe und dem darin nachgewiesenen Insektizid Clothianidin.
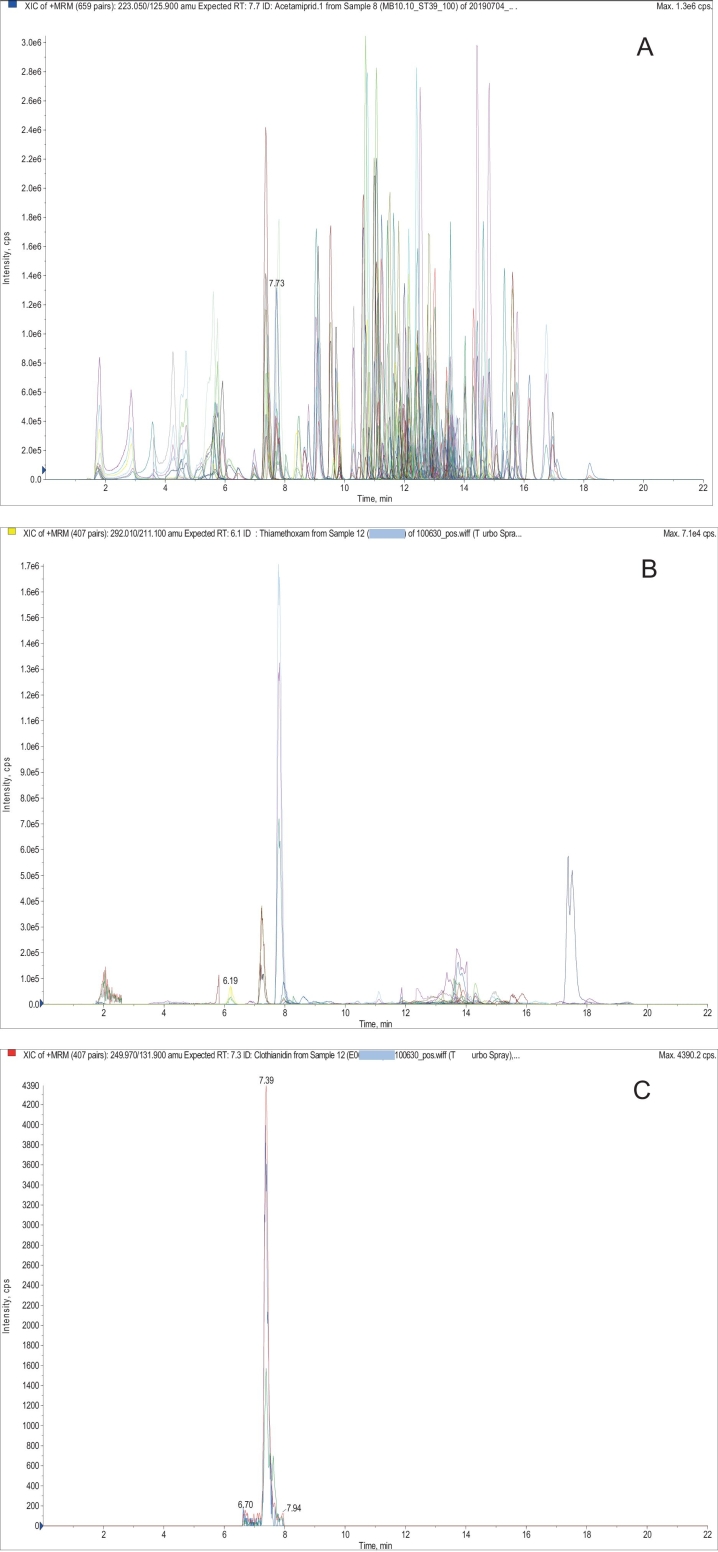
Abb. 3. LC-MS/MS-Chromatogramme eines 100 pg/μl Standards mit 216 Referenzsubstanzen und 5 internen Standards in Bienenmatrix für die Identifizierung und Quantifizierung von u.a. Pflanzenschutzmittelwirkstoffen (A), einer Bienenprobe (B) und dem darin nachgewiesenem Clothianidin in einer vergrößerten Ansicht (C), in der nur die gemessenen für diesen Wirkstoff charakteristischen MRM-Übergänge gezeigt werden (jede bunte Linie entspricht einem Übergang und verbindet die Messpunkte).
Das Multiple Reaction Monitoring (MRM) ist in den meisten Fällen die Standard-Methode für die Bestimmung der Zielsubstanzen, wenn ein Triple-Quadrupol-Massenspektrometer verwendet wird. Im MRM-Modus wird das Molekül-Ion (Precursor‑Ion) des Analyten im ersten Massenfilter (Quadrupol 1) selektiert, durch Kollision mit Stickstoffmolekülen fragmentiert (Quadrupol 2) und drei charakteristische Fragment‑Ionen (Product‑Ions) werden durch einen zweiten Massenfilter (Quadrupol 3) ausgewählt und registriert. Der Haupt‑MRM ist der empfindlichste Übergang (intensivstes Signal) und wird zur Quantifizierung des Analyten (sog. „Quantifier“) verwendet. Mindestens ein weiterer Übergang (MRM) wird zur Bestätigung (sog. „Qualifier“) der Identität der Substanz benötigt. Die Verwendung mehrerer Qualifier‑MRM erhöht die Sicherheit des Nachweises.
Die Rückstände in den Proben werden mit Matrix-Standards quantifiziert. Die dafür benötigten Extrakte werden aus vergleichbarem möglichst rückstandsfreiem Probenmaterial hergestellt und mit den Referenzsubstanzen sowie den deuterierten internen Standards versetzt. Im Rahmen von Projekten können das Proben der unbehandelten Kontrollen sein.
Im Rahmen der Schadensfallklärung werden Standardreihen mit den Matrices „Bienen“ und „Raps“ und den Konzentrationen 0,5, 1, 5, 10, 100 und 500 pg/μl in Acetonitril verwendet. Die Wahl des Lösungsmittels ermöglicht eine Messung mit GC‑MS und LC‑MS/MS aus einem Extrakt. Die Quantifizierung erfolgt über die relativen Peakflächen nach der Methode des internen Standards (SANTE, 2019, ChemgaPedia). Dazu wird eine Kalibrierfunktion aus den Mess-Signalen der oben genannten Standardlösungen, bestehend aus internen Standards und Analyten in bekannten Konzentrationen, erstellt. Die Standardlösungen (Verdünnungsreihe) enthalten alle dieselbe Menge an internen Standards. Die Signale (Peakfläche) beider Komponenten werden für die Berechnung der Kalibrierfunktion (Signalverhältnis Analyt/interner Standard = relative Peakfläche) genutzt.
Im Rahmen von Projekten beinhalten die Kalibrationsreihen entsprechend der Fragestellung und der zu messenden Wirkstoffe in der Regel eine größere Anzahl Matrix-Standards (mindestens 10 Messpunkte) mit Konzentrationen von 0,01 pg/μl bis zu 100 pg/μl.
Müssen die Probenextrakte für eine korrekte Quantifizierung verdünnt werden, können sie ab einer 1:100‑Verdünnung unter Verwendung von Referenzstandards in Lösungsmittel gemessen werden, da die Matrixeffekte durch diese Verdünnung in der Regel ausreichend verringert werden. Bei geringeren Verdünnungen müssen auch die Matrix-Standards entsprechend verdünnt werden, damit die Matrixanteile in Probenextrakten und Standards möglichst gleich sind.
Zwischen den Proben wird Lösungsmittel injiziert und auch die für die Messreihe verwendete Matrix wird, mit und ohne interne Standards, mitgemessen, um Wirkstoff-Blindwerte oder Störungen einzelner MRM-Übergänge und deren Herkunft zu bestimmen.
Methodenüberprüfungen sollen prinzipiell aufzeigen, ob und mit welcher Qualität, die für das komplette Screening ausgewählten Wirkstoffe mit einer Multimethode nachgewiesen werden können. Zum dem Zweck werden definierte Mengen der zu bestimmenden Substanzen den Probenmaterialien zugesetzt und die Analysen in Anlehnung an die Leitlinie der EU mit Hinweisen für Laboratorien zur Validierung und Qualitätssicherung (SANCO bis 2013, seitdem SANTE, in der jeweils aktuellen Version) durchgeführt. Diese sind im „guidance document on analytical quality control and method validation procedures for pesticide residues and analysis in food and feed“ detailliert beschrieben. Kriterien für die Methodenbeurteilung sind die Wiederfindungsraten, Nachweis- (LOD = Limit of Detection) und Bestimmungsgrenzen (LOQ = Limit of Quantification) der gesuchten Substanzen.
Die Zusatzversuche mit den diversen relevanten Probenmaterialien (Bienen, Pflanzen, Pollen, Honig, Gelée Royal, Wachs, Boden) wurden mit dem jeweils aktuellen Wirkstoffspektrum der Untersuchungsstelle für Bienenvergiftungen, verschiedenen Zusatzkonzentrationen und, in der Regel, jeweils fünf Wiederholungen durchgeführt. Die Zusatzkonzentrationen wurden so gewählt, dass möglichst alle Wirkstoffe des Untersuchungsprogramms mit beiden Messsystemen bestimmt werden konnten. Je nach Matrix lag die Zusatzkonzentration zwischen 10 μg/kg und 50 μg/kg. Das war aufgrund der Heterogenität der Wirkstoffe und der Komplexität der Probenmaterialien mit z.T. stark variierenden Begleitstoffanteilen notwendig. Nur mit den Matrices „Bienen“ und „Raps“ wurde auch ein Zusatzversuch mit 1 μg/kg durchgeführt. Ziel war es, die Grenzen der Nachweisbarkeit für die im Fokus des Screenings stehenden Stoffe mit der eingesetzten Methode zu ermitteln.
Diesem Aspekt wird im Rahmen von Projekten durch Zusatzversuche mit den für die Studie ausgewählten Wirkstoffen und – so weit möglich – allen relevanten Probenmaterialien (unbehandelt, wirkstofffreie Kontrollproben) nachgegangen. Die Zusatzkonzentrationen zur Bestimmung der Wiederfindungsraten werden so gewählt, dass sie im Bereich von Nachweis- und Bestimmungsgrenze und relevanter, durchschnittlicher Konzentrationen liegen.
Die Festlegung der LOD-Werte erfolgt anhand der Messergebnisse von Matrix-Standards. Dafür werden fein abgestufte Standardreihen verwendet, die mit den Probenserien mehrfach gemessen werden, da unterschiedliche Mess-Zeitpunkte und Geräte-Zustände zu unterschiedlichen Ergebnissen führen können.
In der LC‑MS/MS wird die LOD als die niedrigste Konzentration festgelegt, bei der mindestens zwei MRM nachgewiesen werden, deren Signale mindestens dreimal höher sind als das Grundrauschen des Chromatogramms und deren Verhältnis im Bereich der geforderten Kriterien liegt (SANTE, 2019). In der GC‑MS ist die LOD die niedrigste Konzentration, bei der mindestens zwei charakteristische Fragment‑Ionen nachgewiesen werden und die Intensität der beiden Fragmente dreimal höher ist als das Grundrauschen des Chromatogramms (s. Abb. 2 B).
Gemäß den Anforderungen der oben genannten SANTE-Leitlinie ist die LOQ die niedrigste Zusatzkonzentration der Validierung, bei der nach Anwendung der vollständigen Methode für alle Analyten mittlere Wiederfindungsraten im Bereich von 70–120% mit einer relativen Standardabweichung (RSD = relative standard deviation) von ≤ 20% erzielt werden.
Wenn es nicht möglich ist, eine ausreichende Anzahl von Validierungsversuchen durchzuführen, um diesem Konzept zu folgen, wird die nächsthöhere Konzentration der Kalibrierungsstandards über der Nachweisgrenze als LOQ festgelegt.
Die Ergebnisse der Validierungen zeigen die Qualität der Multimethode zu dem Zeitpunkt der Überprüfung mit der jeweiligen Probenmatrix und dem aktuellen Zustand der Messgeräte.
In Tab. 2 sind beispielhaft die Ergebnisse für ausgewählte Insektizide, Akarizide und Varroazide zusammengestellt, die seit 2008 unter anderem in den Proben der Untersuchungsstelle nachgewiesen wurden. Bei den Zusatzkonzentrationen 10 μg/kg und 50 μg/kg wurden mit wenigen Ausnahmen Wiederfindungsraten zwischen 70% und 105% mit RSD von 1% bis 12% erreicht. Bei der Zusatzkonzentration 1 μg/kg waren von den 27 Wirkstoffen sieben nicht mehr nachweisbar, wobei es sich durchweg um mit GC‑MS bestimmbare Wirkstoffe handelt. Das als Biozid (Insektensprays) eingesetzte Permethrin ist z.B. erst ab Konzentrationen über 30 μg/kg bestimmbar.
Tab. 2. Wiederfindungsraten, Nachweis- und Bestimmungsgrenzen für ausgewählte Insektizide, Akarizide und Varroazide in Bienenmatrix bei verschiedenen zugesetzten Wirkstoffkonzentrationen
Wirkstoff | Methode | LOD | LOQ | Zusatzkonzentration (n = 4) | |||||||
(μg/kg) | (μg/kg) | 1 μg/kg |
| 10 μg/kg |
| 50 μg/kg | |||||
|
|
|
| WFR % | RSD % |
| WFR % | RSD % |
| WFR % | RSD % |
Acetamiprid | LC | 0,03 | 0,06 | 62 | 12 |
| 95 | 2 |
| 91 | 6 |
Alpha-Cypermethrin | GC | 3,13 | 6,25 | n.n. | n.n. |
| 82 | 3 |
| 76 | 5 |
Beta-Cyfluthrin | GC | 0,63 | 1,25 | 92 | 11 |
| 101 | 11 |
| 77 | 3 |
Brompropylat | GC | 3,13 | 6,25 | n.n. | n.n. |
| 88 | 7 |
| 86 | 4 |
Chlorpyrifos | GC | 0,63 | 1,25 | 94 | 5 |
| 82 | 8 |
| 78 | 12 |
Clothianidin | LC | 0,31 | 0,63 | 103 | 12 |
| 95 | 4 |
| 88 | 7 |
Coumaphos | GC | 3,13 | 6,25 | n.n. | n.n. |
| 92 | 5 |
| 79 | 3 |
Deltamethrin | GC | 6,25 | 12,5 | n.n. | n.n. |
| 101 | 11 |
| 77 | 3 |
Dimethoat | LC | 0,03 | 0,06 | 85 | 3 |
| 101 | 2 |
| 96 | 3 |
Etofenprox | LC | 0,03 | 0,06 | 46 | 7 |
| 72 | 4 |
| 71 | 7 |
Fipronil | GC | 0,63 | 1,25 | 103 | 7 |
| 71 | 11 |
| 76 | 8 |
Imidacloprid | LC | 0,31 | 0,63 | 77 | 7 |
| 97 | 3 |
| 91 | 5 |
Indoxacarb | LC | 0,31 | 0,63 | 48 | 14 |
| 70 | 7 |
| 71 | 5 |
Lambda-Cyhalothrin | GC | 0,63 | 1,25 | 94 | 4 |
| 84 | 3 |
| 74 | 3 |
Omethoat | LC | 0,13 | 0,31 | 94 | 6 |
| 103 | 3 |
| 105 | 2 |
Parathion-methyl | GC | 3,13 | 6,25 | n.n. | n.n. |
| 102 | 7 |
| 88 | 12 |
Permethrin | GC | 31,3 | 43,8 | n.n. | n.n. |
| n.n. | n.n. |
| 94 | 4 |
Phosalon | LC | 0,06 | 0,13 | 55 | 22 |
| 80 | 3 |
| 78 | 5 |
Prallethrin | GC | 0,63 | 1,25 | 69 | 9 |
| 82 | 9 |
| 79 | 12 |
Pirimicarb | LC | 0,01 | 0,03 | 80 | 2 |
| 95 | 1 |
| 93 | 6 |
Propoxur | LC | 0,06 | 0,13 | 79 | 4 |
| 82 | 3 |
| 85 | 9 |
Spinosyn A | LC | 0,06 | 0,13 | 42 | 14 |
| 68 | 5 |
| 64 | 7 |
Spirodiclofen | GC | 3,13 | 6,25 | n.n. | n.n. |
| 93 | 10 |
| 84 | 11 |
Tau-Fluvalinat | GC | 0,63 | 1,25 | 45 | 19 |
| 82 | 5 |
| 74 | 3 |
Tetramethrin | LC | 0,63 | 1,25 | 51 | 26 |
| 79 | 6 |
| 79 | 2 |
Thiacloprid | LC | 0,03 | 0,06 | 55 | 9 |
| 102 | 4 |
| 86 | 8 |
Thiamethoxam | LC | 0,03 | 0,06 | 66 | 9 |
| 104 | 6 |
| 88 | 1 |
LOD = Nachweisgrenze, LOQ = Bestimmungsgrenze, WFR = Wiederfindungsrate, |
| ||||||||||
Für zehn weitere Substanzen wurden Wiederfindungsraten zwischen 42% und 69% erreicht, in zwei Fällen liegt die RSD über 20%. Das zeigt den Einfluss der Gesamtanalyse bei geringen Wirkstoffgehalten in der Bienenmatrix trotz sehr guter Nachweisempfindlichkeit des Messsystems. Für den Wirkstoff Spinosyn A wurden daher auch bei höheren Zusatzkonzentrationen Wiederfindungsraten unter 70% ermittelt (Tab. 2).
Bei den LOD- und LOQ-Werten zeigen sich sehr große Unterschiede bezüglich der Messsysteme. Mit LC‑MS/MS können in der Regel auch sehr geringe Wirkstoffgehalte noch empfindlich gemessen werden. Die berechneten Werte für die LOQ liegen z.T. über der jeweiligen Zusatzkonzentration, für die jedoch Ergebnisse erzielt wurden, die den Kriterien der SANTE-Leitlinie entsprechen. Beim Zusatz mit 1 μg/kg betrifft das etliche mit GC‑MS quantifizierte Wirkstoffe, wie z.B. Chlorpyrifos und beim Zusatz mit 10 μg/kg nur Deltamethrin. Das liegt an den der Berechnung zugrundeliegenden Werten für Einwaage, Aliquotierung und Endvolumen.
Unter Berücksichtigung aller 240 Wirkstoffe, die 2011 im Zusatzversuch mit Bienenmatrix enthalten waren, lässt sich zusammenfassend sagen, dass die Wiederfindungsraten beim Zusatzniveau von 10 μg/kg überwiegend zwischen 70% und 110%, mit relativen Standardabweichungen unter 15% lagen. Für 10% der Substanzen wurden Ergebnisse unter 70% erzielt und 8% nicht nachgewiesen. Letzteres betraf Wirkstoffe, die entweder erst ab höheren Konzentrationen bestimmbar sind (vor allem Herbizide wie z.B. Ethofumesat oder das Akarizid Permethrin), Extramethoden erfordern (z.B. Dithianon, Flumethrin) oder deren Nachweis aufgrund der Instabilität des Wirkstoffs im Probenextrakt (z.B. Amitraz, Prothioconazol) bzw. bei der Messung (z.B. Captan) nicht möglich ist. Diese instabilen Wirkstoffe sind alternativ anhand gebildeter Abbauprodukte nachweisbar. Sehr positiv ist zu bewerten, dass mit der Rückstandsmethode ohne weitere Reinigungsschritte keine Wiederfindungsraten über 120% ermittelt wurden.
Jeder Zusatzversuch dient dazu, das Potential und die Grenzen einer Methode unter den gewählten Bedingungen für die Rückstandsanalyse eines Probenmaterials auszuloten. Um die Anwendbarkeit und Qualität der Multimethode in einem anderen Zusammenhang aufzuzeigen, sind in Abb. 4 die Ergebnisse für 22 ausgewählte Fungizide aus einem Zusatzversuch mit Pollen dargestellt. Die Wiederfindungsraten liegen bei der zugesetzten Konzentration von 20 μg/kg zwischen 76% (Trifloxystrobin) und 106% (Zoxamide) mit relativen Standardabweichungen von 3% bis 17%.
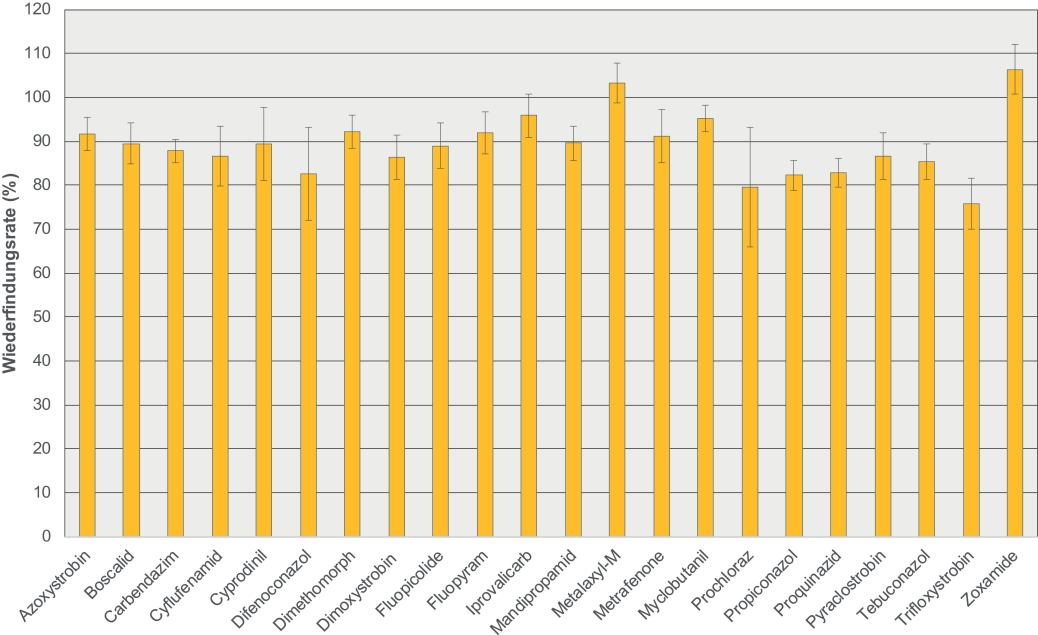
Abb. 4. Wiederfindungsraten für ausgewählte Fungizide in Pollenmatrix bei einer zugesetzten Wirkstoffkonzentration von 20 μg/kg. Die Ergebnisse sind die Mittelwerte aus 5 Wiederholungen, die jeweils zweifach injiziert wurden, mit der absoluten Standardabweichung (± %).
Es ist nicht möglich, mit einer Multimethode alle Analyten mit gleicher Qualität und Empfindlichkeit zu bestimmen. Das gilt generell auch für andere Multimethoden, die für die Bestimmung einer Vielzahl von Pflanzenschutzmittelwirkstoffen mit GC‑MS und/oder LC‑MS/MS in Bienen eingesetzt werden (Walorczyk und Gnuskowski, 2009, Wiest et al., 2011, Kasiotis et al., 2014, Kiljanek et al., 2016).
Die umfänglichen Basisvalidierungen mit allen relevanten Wirkstoffen werden regelmäßig aber aus Kapazitätsgründen nur in größeren zeitlichen Abständen wiederholt. Bei Bedarf erfolgen Überprüfungen der Methode mit neu ins Screening-Programm aufgenommenen Wirkstoffen.
Die Matrix-Standards für die Rückstandsanalysen von Pflanzenmaterial im Rahmen der Schadensfallklärung werden nur mit Raps-Matrix hergestellt. Das liegt darin begründet, dass im Falle von Bienenvergiftungen sehr unterschiedliche Pflanzenproben (Raps-, Kartoffel-, Spargelkraut, Blühstreifenmischung, Blätter von Obstbäumen oder Beerensträuchern) mitgeschickt werden, für die kein unbehandeltes Material für die Herstellung von Matrix-Standards vorliegt. Zusatzversuche mit Raps-, Kartoffel- und Spargelkraut haben gezeigt, dass die Ergebnisse vergleichbar sind und das in diesem Fall auf eine Pflanzen-Matrix zurückgegriffen werden kann, um die Messzeit zu begrenzen und einen möglichst hohen Probendurchsatz zu erreichen.
Wichtig für die Bewertung eines Schadenfalls und die Ermittlung des potentiellen Verursachers des Schadens sind die Identifizierung möglichst vieler übereinstimmender Wirkstoffe in Bienen- und Pflanzenproben und die Konzentrationen der gefundenen toxischen sowie weiterer relevanter Wirkstoffe.
Die Messungen beider chromatographischer Verfahren, GC‑MS und LC‑MS/MS, können starken Matrixeffekten (Signalerhöhung oder ‑unterdrückung) unterliegen, welche die Messung beeinflussen und bei der Quantifizierung zu falschen Ergebnissen führen können. Hajšlová und Zrostlíková haben die Auswirkungen dieser Effekte in der Spurenanalytik und Strategien zu deren Vermeidung bzw. Kompensation bereits 2003 in einem Übersichtsartikel beschrieben. Es müssen also Lösungen gefunden werden, um Fehler zu vermeiden.
Jeder Probe werden zu Beginn der Aufarbeitung deuterierte Wirkstoffe (Surrogat-Standards) für die Qualitätskontrolle der Gesamtanalyse zugesetzt. Die Streuung der Surrogat-Wiederfindungen in Bienenproben aus Schadensfällen gibt einen Einblick in die zum Teil erheblichen Einflüsse der Matrix. Stahnke et al. (2009) fanden, dass für den Großteil der von ihnen untersuchten Wirkstoffe die Eigenschaften der Analyte keinen signifikanten Einfluss auf die Stärke der Signalsuppression in der LC‑MS/MS hatten. Es zeigte sich, dass die Matrixeffekte retentionszeitabhängig waren; da die Retentionszeit bestimmt, mit welcher Fraktion der Matrix die Analyte koeluieren. Eigenschaften und Konzentration der Matrix wurden als entscheidende Parameter für das Ausmaß von Matrixeffekten identifiziert.
Zur Reduktion der Matrixeinflüsse werden in der Bienenanalytik, soweit im Rahmen einer Multimethode möglich, verschiedene Maßnahmen wie Verdünnung der Probenextrakte (Stahnke et al., 2012) und chromatographische Trennung genutzt. Da diese Maßnahmen in der Regel nicht ausreichen, oder wie die Verdünnung nicht immer genutzt werden können, da sehr geringe Mengen für Bienen toxischer Substanzen nachgewiesen werden müssen, wird die Quantifizierung der Wirkstoffe mit Matrix-Standards vorgenommen, um die Matrixeinflüsse zu kompensieren. Um das zu erreichen, müssen Standard und Probe eine sehr ähnliche Zusammensetzung aufweisen.
In Abb. 5 sind Beispiele für die Matrixeffekte bei der Quantifizierung verschiedener Insektizide mit Referenzsubstanzen in Bienenmatrix bzw. Lösungsmittel dargestellt.
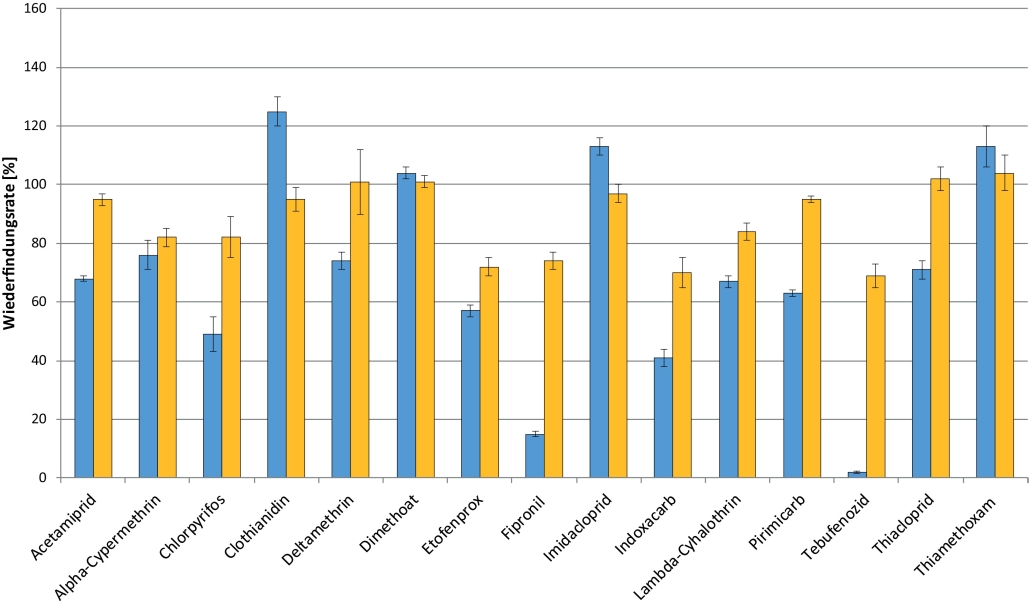
Abb. 5. Matrixeffekte bei der Quantifizierung verschiedener Insektizide in Bienenproben mit Referenzsubstanzen in Lösungsmittel (blaue Säulen) oder Bienenmatrix (gelbe Säulen). Die Zusatzkonzentration war 10 μg/kg. Die Ergebnisse sind die Mittelwerte aus 5 Wiederholungen, die jeweils zweifach injiziert wurden, mit der absoluten Standardabweichung (± %).
Eine weitere Lösung zur Reduktion von Matrixeffekten ist die Verwendung deuterierter (isotopenmarkierter) Referenzsubstanzen als interne Standards für die Quantifizierung. Aufgrund der identischen chemischen Struktur werden für diese internen Standards je nach Messsystem fast die gleichen Retentionszeiten wie für die untersuchten Analyten erreicht. So wird die Messung von internem Standard und Analyt im gleichen Maß durch Matrixkomponenten beeinflusst und die Kalibrierung kann dann mit Referenzsubstanzen in Lösungsmittel durchgeführt werden. Den positiven Effekt zeigen die Beispiele von Dimethoat, Imidacloprid und Alpha-Cypermethrin in Abb. 5, die unter Verwendung von Dimethoat D6, Imidacloprid D4 bzw. Trans-Cypermethrin D6 quantifiziert wurden.
Bei dieser Quantifizierungsmethode sind Isotopenreinheit und Stabilität des isotopenmarkierten Standards unbedingt zu beachtende Faktoren. Selbst bei einer Isotopenreinheit von 99,9% kann der Anteil der nicht markierten, nativen Substanz aufgrund der Messempfindlichkeit besonders bei niedrigen Konzentrationen die Peakfläche des Analyten signifikant beeinflussen bzw. seine Anwesenheit vortäuschen.
Für die Multimethode ist das leider keine allgemein anwendbare Lösung, da nicht für alle Analyten deuterierte „Zwillingssubstanzen“ verfügbar sind. Wenn möglich werden strukturähnliche, überwiegend deuterierte, Substanzen für bestimmte Gruppen (z.B. Pyrethroide, Azol-Fungizide) als interne Standards verwendet. Es sollte in jedem Fall beachtet werden, dass die betreffenden Substanzen so zeitgleich wie möglich eluieren, um eine maximale Kompensation der Matrixeffekte zu erreichen.
In Projekten mit wenigen Zielsubstanzen und verfügbaren deuterierten internen Standards kann von dieser Variante Gebrauch gemacht werden. In Abb. 6 werden am Beispiel des Insektizids Thiacloprid die sehr unterschiedlich ausgeprägten Matrixeffekte in verschiedenen Probenmaterialien im Vergleich mit einem Lösungsmittelstandard aufgezeigt.
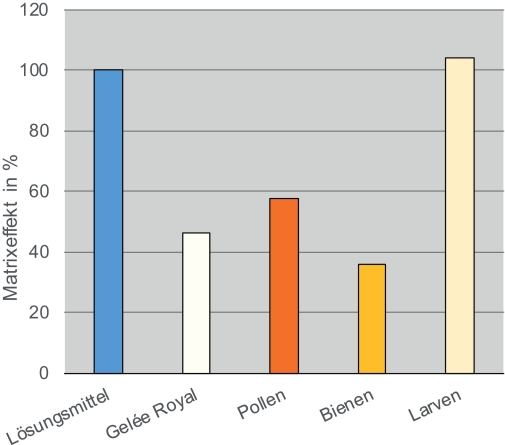
Abb. 6. Matrixeffekte bei der Bestimmung von Thiacloprid in verschiedenen Probentypen im Vergleich zum Lösungsmittel-Standard (Konzentration: 10 pg/μl). Das Ergebnis der Division der Peakfläche des Analyten Thiacloprid durch die Peakfläche des strukturell ähnlichen internen Standards Imidacloprid D4 (= relative Fläche) in Lösungsmittel wurde auf 100% gesetzt. Die Unterschiede zeigen den durch die Begleitstoffe bedingten Matrixeffekt, aus dem in der Regel eine Signalunterdrückung resultiert.
Abbildung 7 zeigt den positiven Effekt des Einsatzes von Thiacloprid D4 als internem Standard bei der Quantifizierung von Thiacloprid in Gelée Royal, Nektar und Pollen. Die mit Matrix- und Lösungsmittel-Standards erzielten Ergebnisse stimmen sehr gut überein, was die sehr weitgehende Kompensation des Matrixeffekts belegt.
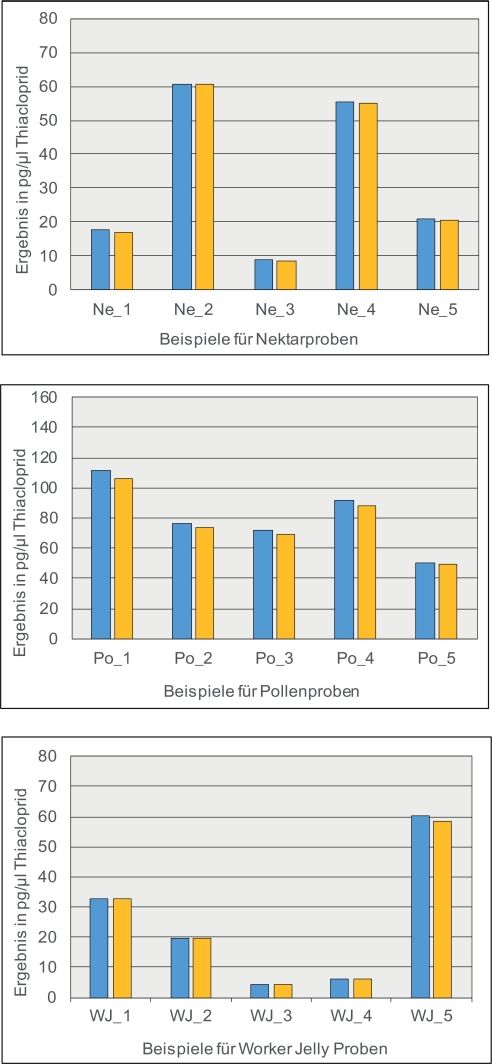
Abb. 7. Kompensation des Matrixeffekts bei der Quantifizierung von Thiacloprid in verschiedenen Probentypen mit Lösungsmittel-Standards (blaue Säulen) im Vergleich zu Matrix-Standards (gelbe Säulen) jeweils unter Verwendung von Thiacloprid D4 als internem Standard. Für die jeweilige Matrix (Nektar, Pollen, Worker Jelly) wurden reale Proben mit Konzentrationen zwischen 4 pg/μl und etwa 100 pg/μl ausgewählt. Die Angaben sind Mittelwerte aus Doppelinjektionen.
Die hier vorgestellte, umfassend validierte Multimethode ist für die Bestimmung von Rückständen in Bienen- und Pflanzenproben sehr gut geeignet, was sich an den erzielten Wiederfindungsraten, LOD- und LOQ-Werten zeigt. Sie ist darüber hinaus vielseitig anwendbar. So waren bei den Bienenprodukten (Pollen/Bienenbrot, Gelée Royal oder Wachs) nur geringfügige Anpassungen (Extraktionsvolumina, Aliquotierung, Endvolumen) erforderlich, um die Multimethode erfolgreich einzusetzen und auch bei geringen Probenmengen ausgewählte Wirkstoffe im unteren Spurenbereich (< 1 μg/kg) bestimmen zu können. Somit ist sowohl eine solide Aufklärung von potentiellen Bienenvergiftungen als auch eine breite Anwendung im Bereich der Forschung möglich.
Jüngste Studien nutzten die Multimethode zur Bestimmung der Rückstandsbelastung essentieller Nahrungsquellen der Honigbienen und deren Auswirkung (Böhme et al., 2017, 2018B). So wurde z.B. der Frage nachgegangen, ob wirkstoffhaltiges Pollenfutter zu einer Kontamination von Königinnen- oder Arbeiterinnen-Futtersaft führt (Böhme et al., 2018a, 2019). Auch die Stabilität von Imidacloprid in Bienen war unter verschiedenen Aspekten nach Fütterung mit einer insektizidhaltigen Zuckerlösung Gegenstand einer Untersuchung (Schott et al., 2017).
Aus der Kombination von Routineanalytik bei der Schadensfallklärung und einem breiten Spektrum experimenteller Fragestellungen resultieren zusätzliche Validierungen der Methode hinsichtlich diverser Matrices, Substanzkombinationen und Konzentrationen.
Wir danken Herrn Dr. Klaus Wallner von der Landesanstalt für Bienenkunde der Universität Hohenheim für die Durchsicht des Manuskripts und die wertvollen Hinweise.
Die Autoren erklären, dass keine Interessenskonflikte vorliegen.
Al-Alam, J., Z. Fajloun, A. Chbani, M. Millet, 2017: A multiresidue method for the analysis of 90 pesticides, 16 PAHs, and 22 PCBs in honey using QuEChERS–SPME. Analytical and Bioanalytical Chemistry 409, 5157–5169, DOI: 10.1007/s00216-017-0463-y.
Al Naggar, Y., G. Codling, A. Vogt, E. Naiem, M. Mona, A. Seif, J.P. Giesy, 2015: Organophosphorus insecticides in honey, pollen and bees (ApismelliferaL.) and their potential hazard to bee colonies in Egypt Yahya. Ecotoxicology and Environmental Safety 114, 1-8, DOI: 10.1016/j.ecoenv.2014.12.039.
Anastassiades, M., S.J. Lehotay, D. Stajnbaher, F.J. Schenck, 2003: Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. Journal of AOAC International 86 (2), 412-431.
Böhme, F., G. Bischoff, C.P.W. Zebitz, P. Rosenkranz, K. Wallner, 2017: Chronic exposure of honeybees, Apis mellifera (Hymenoptera: Apidae) to a pesticide mixture in realistic field exposure rates. Apidologie 48, 353-363, DOI: 10.1007/s13592-016-0479-x.
Böhme, F., G. Bischoff, C.P.W. Zebitz, P. Rosenkranz, K. Wallner, 2018a: From field to food — will pesticide-contaminated pollen diet lead to a contamination of royal jelly? Apidologie 49 (1), 112-119, DOI: 10.1007/s13592-017-0533-3.
Böhme, F., G. Bischoff, C.P.W. Zebitz, P. Rosenkranz, K. Wallner, 2018b: Pesticide residue survey of pollen loads collected by honeybees (Apis mellifera) in daily intervals at three agricultural sites in South Germany. PLOS ONE 13, e0199995, DOI: 10.1371/journal.pone.0199995.
Böhme, F., G. Bischoff, C.P.W. Zebitz, P. Rosenkranz, K. Wallner, 2019: From field to food II — will pesticide-contaminated pollen diet lead to a contamination of worker jelly? Journal of Apicultural Research 58 (4), 112-119, DOI: 10.1080/00218839.2019.1614727.
Calatayud-Vernich, P., F. Calatayud, E. Simó, M.M. Suarez-Varela, Y. Picó, 2016: Influence of pesticide use in fruit orchards during blooming on honeybee mortality in 4 experimental apiaries. Science of the Total Environment 541, 33–41, DOI: 10.1016/j.scitotenv.2015.08.131.
ChemgaPedia, 2020: Chemie, Analytische Chemie, Analytik, Kalibrierung. URL: http://www.chemgapedia.de/vsengine/vlu/vsc/de/ch/3/anc/croma/kalibrierung.vlu/Page/vsc/de/ch/3/anc/croma/datenauswertung/quantitativ/innererstandard/innerstandardm80ht0801.vscml.html
Gil García, M.D., S. Uclés Duque, A.B. Lozano Fernández, A. Sosa, A.R. Fernández-Alba, 2017: Multiresidue method for trace pesticide analysis in honeybee wax comb by GC-QqQ-MS. Talanta 163, 54–64, DOI: 10.1016/j.talanta.2016.10.083.
Goulson, D., E. Nicholls, C. Botias, E.L. Rotheray, 2015: Bee declines driven by combined stress from parasites, pesticides, and lack of flowers. Science 347 (6229), article number 1255957, DOI: 10.1126/science.1255957.
Hajšlová, J., J. Zrostlíková, 2003: Matrix effects in (ultra)trace analysis of pesticide residues in food and biotic matrices. Journal of Chromatography A. 1000 (1-2), 181-197, DOI: 10.1016/S0021-9673(03)00539-9.
IUPAC, 2020: Compendium of Chemical Terminology (the “Gold Book”), URL: http://goldbook.iupac.org/terms/view/M03759.
Kasiotis, K.M., C. Anagnostopoulos, P. Anastasiadou, K. Machera, 2014: Pesticide residues in honeybees, honey and bee pollen by LC-MS/MS screening: Reported death incidents in honeybees. Science of the Total Environment 485, 633-642, DOI: 10.1016/j.scitotenv.2014.03.042.
Kiljanek, T., A. Niewiadowska, S. Semeniuk, M. Gaweł, M. Borzęcka, A. Posyniak, 2016: Multi-residue method for the determination of pesticides and pesticide metabolites in honeybees by liquid and gas chromatography coupled with tandem mass spectrometry — Honeybee poisoning incidents. Journal of Chromatography A. 1435, 100-114, DOI: 10.1016/j.chroma.2016.01.045.
Klein, J., L. Alder, 2003: Applicability of Gradient Liquid Chromatography with Tandem Mass Spectrometry to the Simultaneous Screening for About 100 Pesticides in Crops. Journal of AOAC International 86 (5), 1015-1037, DOI: 10.1093/jaoac/86.5.1015.
Pistorius, J., 2016: Vergiftungen von Honigbienen (Apis mellifera L.) durch insektizidhaltigen Staubabrieb beim Anbau von Raps und Mais. Dissertation, Universität Rostock, Dissertationen aus dem Julius Kühn-Institut,188 S., DOI: 10.5073/dissjki.2016.003.
Potts, S.G., J.C. Biesmeijer, C. Kremen, P. Neumann, O. Schweiger, W.E. Kunin, 2010: Global pollinator declines: trends, impacts and drivers. Trends in ecology & evolution 25 (6), 345–353, DOI: 10.1016/j.tree.2010.01.007.
Przybylski, C., C. Segard, 2009: Method for routine screening of pesticides and metabolites in meat based baby-food using extraction and gas chromatography-massspectrometry. Journal of Separation Science 32, 1858–1867, DOI: 10.1002/jssc.200900016.
Roszko, M.L.M. Kamińska, K. Szymczyk, R. Jedrzejczak:, 2016: Levels of selected persistent organic pollutants (PCB, PBDE) and pesticides in honey bee pollen sampled in Poland. PLOS ONE 11, e0167487, DOI: 10.1371/journal.pone.0167487.
SANTE, 2019: XXX/12682/2019 (Implemented by 01.01.2020): Method Validation and Quality Control Procedures for Pesticide Residues Analysis in Food and Feed, URL: https://www.eurl-pesticides.eu/docs/public/tmplt_article.asp?CntID=727.
Schott, M., G. Bischoff, G. Eichner, A. Vilcinskas, R. Büchler, M.D. Meixner, A. Brandt, 2017: Temporal dynamics of whole body residues of the neonicotinoid insecticide imidacloprid in live or dead honeybees. Scientific reports 7, Article number: 6288, DOI: 10.1038/s41598-017-06259-z.
Seefeld, F., 2008: Chemische Untersuchungen zur Aufklärung von Schadensfällen an Honigbienen durch Pflanzenschutzmittel im Zeitraum 1985 bis 2006. Mitteilungen aus dem Julius Kühn-Institut Heft 418.
Stahnke, H., T. Reemtsma, L. Alder, 2009: Compensation of Matrix Effects by Postcolumn Infusion of a Monitor Substance in Multiresidue Analysis with LC-MS/MS. Analytical Chemistry 81 (6), 2185-2192, DOI: 10.1021/ac802362 s.
Stahnke, H., S. Kittlaus, G. Kempe, L. Alder, 2012: Reduction of matrix effects in liquid chromatography-electrospray ionization-mass spectrometry by dilution of the sample extracts: How much dilution is needed? Analytical Chemistry 84 (3), 1474-1482. DOI: 10.1021/ac202661j.
Tette, P.A.S., F.A. Da Silva Oliveira, E.N.C. Pereira, G. Silva, M.B. De Abreu Glória, C. Fernandes, 2016: Multiclass method for pesticides quantification in honey by means of modified QuEChERS and UHPLC-MS/MS. Food Chemistry 211, 130-139, DOI: 10.1016/j.foodchem.2016.05.036.
Vanbergen, A.J., S.G. Potts, A. Vian, E.P. Malkemper, J. Young, T. Tscheulin, 2019: Risk to pollinators from anthropogenic electro-magnetic radiation (EMR): Evidence and knowledge gaps. Science of the Total Environment 695, Article number 133833, DOI: 10.1016/j.scitotenv.2019.133833.
Walorczyk, S., B. Gnuskowski, 2009: Development and validation of a multi-residue method for the determination of pesticides in honeybees using acetonitrile-based extraction and gaschromatography–tandem quadrupole mass spectrometry. Journal of Chromatography A. 1216, 6522-6531, DOI: 10.1016/j.chroma.2009.07.045.
Wernecke, A., M. Frommberger, R. Forster, J. Pistorius, 2019: Letale Auswirkungen verschiedener Tankmischungen aus Insektiziden, Fungiziden und Düngemitteln auf Honigbienen unter Labor-, Halbfreiland- und Freilandbedingungen. Journal of Consumer Protection and Food Safety 14, 239–249, DOI: 10.1007/s00003-019-01233-5.
Wiest, L., A. Buleté, B. Giroud, C. Fratta, S. Amic, O. Lambert et al., 2011: Multi-residue analysis of 80 environmental contaminants in honeys, honeybees and pollens by one extraction procedure followed by liquid and gas chromatography coupled with mass spectrometric detection. Journal of Chromatography A; 1218 (34), 5743–5756, DOI: 10.1016/j.chroma.2011.06.079.
Wisk, J.D., 2014: Assessing Exposure of Pesticides to Bees. In: Pesticide Risk Assessment for Pollinators. Fischer, D., T. Moriarty (Hrsg.), Wiley Blackwell, 45-74, DOI: 10.1002/9781118852408.ch7.

 Suchen
Suchen