
Journal für Kulturpflanzen, 74 (11-12). S. 282–286, 2022 | DOI: 10.5073/JfK.2022.11-12.08 | Jung
Induced mutations for studying Mendelian inheritance
Induzierte Mutationen zum Studium der Mendelschen Vererbung
 | (c) The author(s) 2022 This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/deed.en). |
Submitted/accepted for publication: 9 August 2022/3 November 2022 |
All genetic variation results from mutations. Orders of magnitude can increase mutation rates by applying irradiation or chemical treatment. Genome editing offers new perspectives for mutation induction because mutation sites can be precisely targeted even within large plant genomes for the first time. Generally, transgenes after genetic engineering also result in new genetic variation. All single-gene mutations after genome editing, transgenesis, or chemical mutagenesis are inherited according to the Mendelian rules. Thus, offspring can be phenotypically classified into discrete classes, whereas polygenic inheritance results in continuous variation. However, genome-wide studies have blurred the boundaries between the two in recent years. Single-gene knockout mutations can be inherited non-Mendelian, mainly if transcription factor genes are targeted. Even classical Mendelian traits are now believed to be controlled by numerous genes. Therefore, Mendelian genetics should be limited to genetic variation, whereas phenotypic variation should be considered polygenic.
mutagenesis, CRISPR-Cas, genome editing, TILLING, TILLING by sequencing, polyploidy
Sämtliche genetische Variation resultiert aus Mutationen. Die Mutationsraten können durch Bestrahlung oder chemische Behandlung um mehrere Größenordnungen erhöht werden. Die Genom-Editierung bietet neue Perspektiven für die Mutationsinduktion, da erstmals Zielsequenzen auch innerhalb großer Pflanzengenome präzise anvisiert werden können. Im weitesten Sinne sind auch Transgene, die mit gentechnischen Verfahren in ein Genom eingeschleust wurden, Mutationen, die zu neuer genetischer Variation führen. Alle Einzelgen-Mutationen nach Genom-Editierung, Transgenese oder chemischer Mutagenese werden nach den Mendelschen Regeln vererbt. Die Nachkommen können phänotypisch in diskrete Klassen eingeteilt werden, während polygene Vererbung zu kontinuierlicher Variation führt. Aber aufgrund genomweiter Studien ist eine derartige Einteilung nicht mehr gerechtfertigt. Knockout-Mutationen einzelner Gene können auf eine nicht-mendelnde Weise vererbt werden, vor allem, wenn Gene mutagen verändert werden, die für Transkriptionsfaktoren kodieren. Sogar klassische Mendel-Merkmale werden nach heutiger Kenntnis heute von zahlreichen Genen kontrolliert. Daher sollte die Mendelsche Genetik auf die genetische Variation beschränkt werden, während die phänotypische Variation als polygen angesehen werden sollte.
Mutationen, Mutagenese, CRISPR-Cas, genome editing, TILLING, TILLING by sequencing, Polyploidie
All genetic variation is caused by mutations that occur randomly and spontaneously. There are different types of mutations, ploidy mutations affecting whole genomes, chromosome mutations such as insertions/deletions, translocations, and inversions, and single-gene or single-nucleotide mutations. Mutations can occur spontaneously and can be induced by chemicals or by irradiation. Thanks to genome research, there is a good understanding of how often mutations occur spontaneously over generations. The frequency of spontaneous natural mutagenesis ranges from 10–8–10–9 per bp (Ossowski et al., 2010; Pixley et al., 2022). In this article, I will refer to induced mutations on the single nucleotide or single-gene level.
Since the early 20th century, it has been known that mutation rates can be drastically increased by gamma irradiation or fast neutron irradiation (Jung & Till, 2021). The mutations induced range from single nucleotide deletions to large chromosome rearrangements. In contrast, chemicals such as ethyl-methanesulfonate (EMS) cause single-nucleotide mutations. Induced mutations have been used in plant breeding for more than 70 years. The number of mutations within a single genome can be enormous. For example, after whole-genome sequencing of a rapeseed EMS mutant population, each M2 plant was found to carry 39,000 mutations on average (unpublished data).
Recently, the mutation paradigm has been changed. Genome editing enables precise mutation induction within a sequence previously selected. The CRISPR-Cas method is frequently applied to induce mutations between one and a few hundred nucleotides (Zhu et al., 2020). Further refinement of this method led to precise base changes (base editing) and sequence incorporation (gene replacement). In a broader context, genetic modification after transformation results in mutations by adding a new gene to a plant genome.
Mutations affecting single genes in a diploid plant follow the Mendelian rules of inheritance. However, a clear distinction should be made between genotypic and phenotypic inheritance. As long as the mutation does not cause lethality or reduced fitness, it is inherited as a new allele. For example, in an M2 family derived from a heterozygous M1 plant, 75% of the plants carry the mutant allele disregarding the chimeric nature of M1 plants (Figure 1).
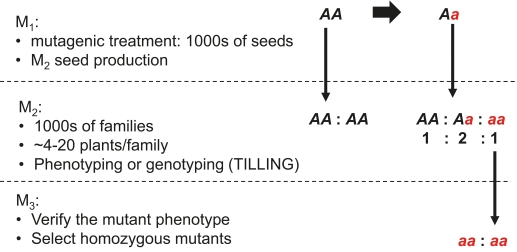
Figure 1. Inheritance of mutant alleles in segregating populations of diploid species.
The Mendelian rules set the foundations for mutant screening in the offspring of mutagenized populations. Because mutations within a target gene a rare, thousands of M2 families need to be screened phenotypically. The mutant allele can be fixed in the next generation, giving rise to a homozygous mutant line. Unfortunately, mutant screening in polyploid plants is much more complicated because two or more copies of a gene exist. In autopolyploids, they are duplicates with essentially the same function. Therefore, the frequency of homozygotes in an M2 population depends on the ploidy level. If one allele has been mutagenized (Am) in an autotetraploid species, completely homozygous plants (quadruplex type, AmAmAmAm) can be found only in the M3 generation. Suppose we can identify the duplex type in the M2 (AmAmAxAx), which is only possible by DNA-based genotyping methods. After selfing this plant, the quadruple mutant in the M3 is expected with a frequency of 2.8%. Here, the CRISPR-Cas technology is superior because we can get a quadruple mutant already in the M1 generation.
In allopolyploid plants, homoeologous gene copies (paralogs) may have undergone neofunctionalization during evolution resulting in altered gene function. Historic duplications within genomes during evolution further complicate genetics. For example, rapeseed (Brassica napus) is an allotetraploid species (2n=4x=38). As a result of subgenome triplication, each gene of the related Brassica species Arabidopsis thaliana is represented by 4-6 copies in B. napus (Chalhoub et al., 2014) which severely complicates phenotypic mutant detection because only multiple mutation events give rise to a new phenotype. Therefore, huge M2 populations must be screened to find a plant with multiple mutation events within the desired genes. For example, in our rapeseed mutant population, a single knockout mutation within a given gene is commonly found among ~500 M1 offspring. For a double gene mutation, the frequency would decrease to 1 out of 250,000 M1 plants demonstrating the difficulty in finding multiple mutations by phenotypic screening.
The situation changed drastically with the advent of sequence-based mutant screening methods commonly referred to as TILLING (Targeted Induced Local Lesions in Genomes) (Jung & Till, 2021), where mutations are detected by sequence analysis and not by phenotypic screening. In this way, mutant pyramiding became possible because single mutations from different M1 plants could be combined by crossing to create multiple mutant genotypes (Braatz et al., 2018; Sashidhar et al., 2020a; Karunarathna et al., 2020). Recently, TILLING-by-sequencing has been applied in wheat (Krasileva et al., 2017) and rapeseed (unpublished data), where the exomes or whole genomes of M2 plants are sequenced. Within a mutant family carrying the desired mutation, the mutant haplotype frequency is 50%. Consequently, sequencing four plants from each M2 family is sufficient to find any mutation with a high probability. Typically, the bulked DNA is sequenced, reducing costs by 75% (unpublished data). In conclusion, Mendelian genetics are fundamental for mutant screening either by phenotype or genotype.
Since the 1980s, gene transfer has become a routine technique for studying gene function and creating new genetic variations in crops. In a broader sense, genetically modified organisms can be regarded as mutants. When the first genetically modified (transgenic) plant was produced in 1983, it was unknown whether the introduced gene was inherited as a Mendelian gene. But soon, it became clear that even transgenes from distantly related species are inherited according to Mendelian rules either as one or >1 gene owing to the number of insertion events once they have been stably incorporated into a host genome. This was the reason for their use in plant breeding because transgenes could be easily introduced to elite lines and selected in offspring generations by standard backcrossing.
In 2012, genome editing (GE) became routine based on the CRISPR-Cas technology. GE caused a shift in paradigm because mutations can be precisely induced at any position in the genome in contrast to Agrobacterium-mediated or vector-free gene transfer, where the integration site cannot be predicted. Like random mutations, mutations after GE are inherited as single Mendelian genes. CRISPR-Cas-induced knockout mutations are recessive and inherited according to Mendelian rules for dominant/recessive inheritance. Therefore, the presence of both mutant and wildtype alleles is called 'heterozygosity' in complete correspondence with Mendelian genetics. In a recent study from our lab, a CRISPR-Cas construct was transformed into rapeseed by Agrobacterium-mediated gene transfer (Table 1) (Braatz et al., 2017). The target gene ALCATRAZ (ALC) is involved in valve margin development and, thus, contributes to seed shattering from mature fruits, which before or during harvest, causes yield loss. Knockouts of two ALC paralogs resulted in increased pod-shattering resistance. In the T2 offspring of a single T1 transformant, the transgene segregated in a 3:1 manner typical for a single transformation event resulting in a hemizygous genotype. In rapeseed, the probability of linkage between transgene and target gene is 1/19. As expected, the transgene was not linked with the two ALC genes referred to as 'A' and 'C' (Table 1) because there were non-transgenic plants with all four mutant alleles. Altogether, we found eight out of nine genotypes expected for random segregation. Thus, non-transgenic mutants could be easily selected in the T2 generation. The two ALC genes segregated in a 1:2:1:2:4:2:1:2:1 manner which follows a digenic inheritance with random segregation.
Table 1. Inheritance of CRISPR-Cas9 induced mutations in two BnALC genes (A, C) of rapeseed. 36 T2 plants were tested for the presence of the transgene and the CRISPR-Cas induced mutations by PCR. O = observed, E = expected number of plants (Braatz et al., 2018).
| Transgene genotypes |
| alc genotypes | |||||||||||
| Trans- | Non-transg. | Chi2 testb |
| A2A2 | A2A2 | A2A2 | A2A3 | A2A3 | A2A3 | A3A3 | A3A3 | A3A3 | Chi2 testc |
O | 27 | 9 | 0 |
| 3 | 4 | 0 | 3 | 14 | 2 | 1 | 6 | 3 | 8.52 |
Ea | 27 | 9 | 2.25 | 4.25 | 2.25 | 4.25 | 9 | 4.25 | 2.25 | 4.25 | 2.25 | |||
a: under the assumption that the T1 parent was non-chimeric (A2A3/C2C3)
b: 3:1 segregation, Chi2(0.999;2) = 13.82
c: 1:2:1:2:4:2:1:2:1 segregation, Chi2(0.999; 8) = 26.12
De novo mutations can complicate Mendelian inheritance after GE in subsequent generations. For example, after the knockout of phytic acid synthesis genes in rapeseed by GE, new mutant alleles appeared in the T2 generation (Sashidhar et al., 2020b). In this case, non-Mendelian inheritance can be explained by the chimeric nature of the T1 plants and the inability of Cas9 to produce double-strand breaks in the initial transformant. If the Cas9 nuclease does not induce stable mutations in the first generation, it continues cleaving the target sequence in the next generation(s), giving rise to new mutant alleles. However, once the mutation has been 'stabilized' due to the absence of the Cas9 gene or the alteration of the target sequence, the new mutant alleles are inherited in a Mendelian way. In conclusion, phenotypic studies on CRISPR-Cas mutagenized plants are only valid if homozygosity of the mutant locus has been proven, which gives rise to lines fixed for the mutant alleles (Jung & Till, 2021).
There are different reasons why mutations do not always show the desired phenotypic effect, even in the case of homozygosity at the target locus. Gene redundancy can mask the mutant locus because functional paralogs can substitute its function in polyploids and diploids undergoing one or more genome duplications during evolution. Moreover, suppose the target gene is part of a highly variable genetic and metabolic pathway. In that case, it can be replaced by other genes encoding enzymes or transcription factors with equal or similar functions. In this respect, flowering time regulation can serve as a prime example. There are major flowering time genes whose gene products are important hubs of flowering regulatory pathways, such as the Arabidopsis FLOWERING LOCUS T (FT) gene which encodes the florigen necessary for the transformation of the shoot apical meristem into a floral meristem (Blümel et al., 2015). Functional orthologs have been identified in most flowering plants studied so far. From the first glimpse, these are typical Mendelian genes with an apparent phenotype. They had been found after random mutagenesis followed by phenotypic screening (Koornneef et al., 1991). But even in the case of a single copy gene, knockout does not eliminate the competence to flower. Mutants can still perform the floral transition, albeit with a significant delay. It is known that other transcription factors and sRNAs can partly substitute FT, masking its knockout mutation's effect and shifting the qualitative (Mendelian) into a (semi)quantitative inheritance. If the phenotypic effect is low, the borders between Mendelian and quantitative inheritance vanish (Fig. 2).
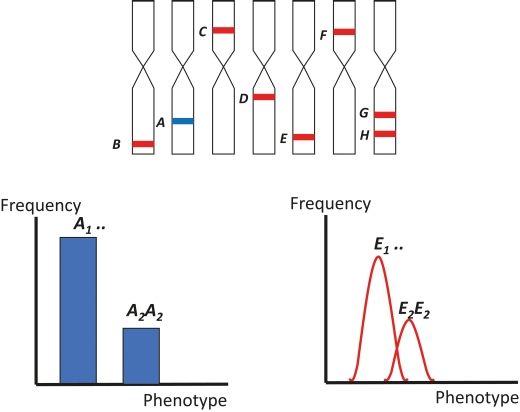
Figure 2. Frequency distributions and phenotypic values in segregating populations with qualitative (blue) and quantitative (red) inheritance. A is a Mendelian gene with complete dominance controlling a phenotype (h2 = 1). E is a major QTL with partial dominance controlling a phenotype together with six other QTLs and depending on the environment (h2<1).
This casts doubt on the concept of Mendelian genetics, as outlined in a recent opinion paper (Tautz et al., 2020). In the past years, many studies revealed that single mutations could cause major changes in global transcription profiles which is in line with the fact that a single transcription factor can control hundreds of downstream targets. Tautz et al. (2020) suggested reassessing the infinitesimal inheritance model, which has been the basis of quantitative genetics since Ronald Fisher worked out its statistical foundations in the 1920s. Today, in contrast to previous approaches, QTLs can be precisely localized, and their phenotypic effects can be calculated (Fig. 2). The knockout of a single gene contributing to a quantitative trait, such as in the case of the transcription factors FT and ALC, does not result in discrete phenotypic classes but increases quantitative variation. In contrast, knocking out a Mendelian gene, such as the Hs4 gene causing complete resistance to the beet cyst nematode (Heterodera schachtii) in sugar beet by a CRISPR-Cas-induced single nucleotide deletion resulted in complete loss of resistance (Kumar et al., 2021). As a result, the offspring of plants heterozygous at the Hs4 locus segregated into two distinct phenotypes classes, resistant and susceptible, which is in line with monogenic Mendelian inheritance.
In the past years, DNA-based mutant studies have confirmed Mendel's rules. The inheritance of mutant alleles follows the Mendelian rules as long as they are not part of major chromosomal rearrangements. However, genome-wide QTL and global transcription studies have revised our view of the phenotypic inheritance of "Mendelian" phenotypes, which can be much more complex. Transcriptome profiling and functional analysis of transcription factor genes shed new light on the complexity of Mendelian inheritance. While Mendel's rules are still valid regarding genotypic inheritance, they need to be revised to understand the makeup of phenotypes even if single genes control them.
This offers new perspectives for breeding traits of agronomic importance, which seemingly are controlled by polygenes but can be improved by only altering the genotype at a single (e.g., transcription factor) locus as has been suggested for improving yield capacity in rice and other cereals (Wang & Zhang, 2017). Therefore, new allelic variation will be needed. Manipulating quantitative traits by single-gene mutagenesis will be a challenge in the future. Gene transfer and CRISPR-Cas-mediated mutagenesis are already routine for many crops. Unfortunately, these technologies are (still) inaccessible to European breeders who can only exploit existing allelic variation or new variation induced by random mutagenesis (Jung & Till, 2021). This comes with several shortcomings, which whole genome sequencing of mutant populations could partly overcome in combination with genomic background screening.
The author declares that he does not have any conflicts of interest.
Blümel, M., N. Dally, C. Jung, 2015: Flowering time regulation in crops — what did we learn from Arabidopsis? Current Opinion in Biotechnology 32, 121-129, DOI: 10.1016/j.copbio.2014.11.023.
Braatz, J., H.-J. Harloff, N. Emrani, C. Elisha, L. Heepe, S.N. Gorb, C. Jung, 2018: The effect of INDEHISCENT point mutations on silique shatter resistance in oilseed rape (Brassica napus). Theoretical and Applied Genetics 131, 959-971, DOI: 10.1007/s00122-018-3051-4.
Braatz, J., H. Harloff, M. Mascher, N. Stein, A. Himmelbach, C. Jung, 2017: CRISPR-Cas9 targeted mutagenesis leads to simultaneous modification of different homoeologous gene copies in polyploid oilseed rape (Brassica napus L.) Plant Physiology 174, 935-942, DOI: 10.1104/pp.17.00426.
Chalhoub, B., F. Denoeud, S. Liu, I.A.P. Parkin, H. Tang, X. Wang, J. Chiquet, H. Belcram, C. Tong, B. Samans, M. Corréa, C. Da Silva, J. Just, C. Falentin, C.S. Koh, I. Le Clainche, M. Bernard, P. Bento, B. Noel, K. Labadie et al., 2014: Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345 (6199), 950-953, DOI: 10.1126/science.1253435.
Jung, C., B. Till, 2021: Mutagenesis and genome editing in crop improvement: perspectives for the global regulatory landscape. Trends in Plant Science 26, 1258-1269, DOI: 10.1016/j.tplants.2021.08.002.
Karunarathna, N.L., H. Wang, H.J. Harloff, L. Jiang, C. Jung, 2020: Elevating seed oil content in a polypoid crop by induced mutations in SEED FATTY ACID REDUCER genes. Plant Biotechnology Journal 18, 2251-2266, DOI: 10.1111/pbi.13381.
Koornneef, M., C.J. Hanhart, d. van, V, 1991: A genetic and physiological analysis of late flowering mutants in Arabidopsis thaliana. Molecular and General Genetics MGG 229 (1), 57-66, DOI: 10.1007/BF00264213.
Krasileva, K.V., H.A. Vasquez-Gross, T. Howell, P. Bailey, F. Paraiso, L. Clissold, J. Simmonds, R.H. Ramirez-Gonzalez, X. Wang, P. Borrill, C. Fosker, S. Ayling, A.L. Phillips, C. Uauy, J. Dubcovsky, 2017: Uncovering hidden variation in polyploid wheat. Proceedings of the National Academy of Sciences 114 (6), E913-E921, DOI: 10.1073/pnas.1619268114.
Kumar, A., H. Harloff, S. Melzer, J. Leineweber, B. Defant, C. Jung, 2021: A rhomboid-like protease gene from an interspecies translocation confers resistance to cyst nematodes New Phytologist 231, 801-813, DOI: 10.1111/nph.17394.
Ossowski, S., K. Schneeberger, J.I. Lucas-Lledó, N. Warthmann, R.M. Clark, R.G. Shaw, D. Weigel, M. Lynch, 2010: The Rate and Molecular Spectrum of Spontaneous Mutations in Arabidopsis thaliana. Science 327 (5961), 92-94, DOI: 10.1126/science.1180677.
Pixley, K.V., J.B. Falck-Zepeda, R.L. Paarlberg, P.W.B. Phillips, I.H. Slamet-Loedin, K.S. Dhugga, H. Campos, N. Gutterson, 2022: Genome-edited crops for improved food security of smallholder farmers. Nature Genetics 54 (4), 364-367, DOI: 10.1038/s41588-022-01046-7.
Sashidhar, N., H. Harloff, C. Jung, 2020a: Identification of phytic acid mutants in oilseed rape (Brassica napus L.) by large scale screening of mutant populations through amplicon sequencing. New Phytologist 225, 2022–2034, DOI: 10.1111/nph.16281.
Sashidhar, N., H.J. Harloff, L. Potgieter, C. Jung, 2020b: Gene editing of three BnITPK genes in tetraploid oilseed rape leads to significant reduction of phytic acid in seeds. Plant Biotechnology Journal 18, 2241-2250, DOI: 10.1111/pbi.13380.
Tautz, D., G. Reeves, L.F. Pallares, 2020: New experimental support for long standing concepts of polygenic genetics implies that the Mendelian genetic paradigm needs to be revised. The New (Old) Genetics. Wittinghofer, A.H. Jäckle, Halle, DOI: 10.34714/leopoldina_nal-live_0001_01000.
Wang, L., Q. Zhang, 2017: Boosting Rice Yield by Fine-Tuning SPL Gene Expression. Trends in Plant Science 22 (8), 643-646, DOI: 10.1016/j.tplants.2017.06.004.
Zhu, H., C. Li, C. Gao, 2020: Applications of CRISPR–Cas in agriculture and plant biotechnology. Nature Reviews Molecular Cell Biology 21 (11), 661-677, DOI: 10.1038/s41580-020-00288-9.

 Suchen
Suchen